Unfassbar selten: Es gibt eine Krankheit, an der nach aktuellem Kenntnisstand nur drei Personen auf dieser Welt erkrankt sind. Auch wenn dies ein Extremfall ist, gibt es von solchen seltenen Erkrankungen insgesamt sehr viele. Deshalb sind weltweit Millionen Menschen betroffen. Trotzdem sind Medikamente und Therapien bisher genauso rar wie die Krankheiten selbst.
Der 29. Februar ist nicht nur der seltenste Tag des Jahres, sondern auch der Tag der seltenen Erkrankungen. Er soll die wenig bekannten Krankheiten, von denen trotzdem so viele Menschen betroffen sind, mehr in den Fokus rücken. Denn die Seltenheit dieser Leiden hat zur Folge, dass nur wenig Geld in die medizinische Forschung investiert wird. Daher steht sie in diesem Bereich noch ganz am Anfang.
Betroffene leiden dadurch meist ihr ganzes Leben an den Symptomen ihrer Krankheit und haben nur geringe Chancen auf Heilung. Oft wird die Krankheit sogar über Jahre nicht erkannt oder falsch diagnostiziert und die Erkrankten müssen mit der Ungewissheit über ihre Symptome leben.
Der lange Weg zur Diagnose
Selten und doch ein Massenphänomen
Selten und doch häufig: Krankheiten von denen man noch nie etwas gehört hat, gibt es häufiger als gedacht. Eine Krankheit gilt in der Europäischen Union als selten, wenn weniger als fünf von 10.000 Menschen davon betroffen sind. Das klingt erstmal wenig, doch die Liste der seltenen Erkrankungen ist lang. Etwa 7.000 seltene Krankheiten sind bekannt, wobei regelmäßig neue Krankheitsbilder beschrieben werden.
In der Summe sind davon weltweit etwa 350 Millionen Menschen betroffen. Somit erreichen die seltenen Erkrankungen in ihrer Gesamtheit ähnlich hohe Fallzahlen wie manche Volkskrankheiten. Trotzdem werden die seltenen Krankheiten oft unterschätzt und sind zum Teil sehr wenig erforscht. Sie werden daher auch als „orphan diseases“ und die Betroffenen manchmal als „Waisenkinder der Medizin“ bezeichnet.
Was aber bedeutet das für Menschen, deren Krankheit als selten eingestuft wird? „Keine Krankheit kann zu selten sein, um ihr Aufmerksamkeit zu schenken“, steht auf der Website des Orphanet – einem Portal, das Informationen über seltene Krankheiten bündelt. Denn umfassende Datenbanken zu diesen Krankheiten gibt es kaum, sodass die seltenen Erkrankungen von Ärzten oft nicht erkannt werden.
Zusätzlich erschwert wird die Diagnose durch die Komplexität der Symptome, welche meist mehrere Organe betreffen. So kann es im Durchschnitt fünf Jahre dauern, bis die Betroffenen die korrekten Befunde erhalten. Auf dem schwierigen Weg zur richtigen Diagnose ihrer Krankheit suchen die Patienten bis zu acht verschiedene Ärzte auf. Mindestens die Hälfte erhält dabei einmal eine Fehldiagnose, da die Symptome oft mit anderen Krankheitsbildern verwechselt werden. Außerdem werden die Patienten zum Teil nicht ernst genommen oder ihnen wird sogar vorgeworfen, sich ihre außergewöhnliche Symptomatik nur auszudenken.
Die Symptome von seltenen Erkrankungen treten oft bereits im Kindesalter auf und die ergebnislose Suche von Arzt zu Arzt ist meist eine zusätzliche seelische Belastung. „Diese Patienten haben eine wahre Odyssee quer durch unser Gesundheitssystem hinter sich“, sagt der Kardiologe Jürgen Schäfer, Leiter des Zentrums für unerkannte und seltene Erkrankungen in Marburg. „Der Leidensdruck der Betroffenen ist enorm“.
Wenn die Erkrankten den langen Ärzte-Marathon zur Diagnose gemeistert haben, warten allerdings weitere Schwierigkeiten auf sie: Von den mehreren tausend Krankheiten mit Seltenheitsstatus sind bisher nur etwa 130 mit in der Europäischen Union zugelassenen Medikamenten behandelbar.
Für manche Krankheiten existieren demnach zwar Behandlungsmöglichkeiten, doch für die meisten Betroffenen liegen spezifische Medikamente in weiter Ferne. Dies liegt schlicht daran, dass so wenige dieser Krankheiten bisher erforscht sind. Zum einen sind die Krankheiten wenig bekannt und deswegen seltener Gegenstand einer Studie. Zum anderen sorgt die Seltenheit der Erkrankungen dafür, dass es häufig zu wenig Studienteilnehmer gibt, an denen die Krankheit erforscht werden kann.
Außerdem liegt die Medikamentenentwicklung für seltene Krankheiten oft nicht im Interesse der Pharmaunternehmen, weil sich der Aufwand von einem wirtschaftlichen Standpunkt aus gesehen „nicht lohnt“. Es würde schlicht zu wenig Abnehmer für ein Arzneimittel geben. Die Behandlung der Symptome ist also oft alles was den Ärzten bleibt.
Seltene Erkrankungen haben verschiedene Ursachen
Mutation oder Infektion?
Die Symptome von seltenen Erkrankungen treten meist schon kurz nach der Geburt auf, da 80 Prozent dieser Leiden genetisch bedingt sind. Häufig sind sie damit chronisch und halten ein Leben lang an. Manche können sogar lebensbedrohlich sein. Doch die Gene sind nicht immer verantwortlich. In einigen Fällen werden die seltenen Krankheiten auch durch Infektionen beispielsweise mit Viren ausgelöst.
Die meisten Ursachen seltener Erkrankungen lassen sich auf nur ein einziges Gen im Erbgut zurückführen. Oft wurde in diesem Gen nur eine der DNA-Basen durch eine sogenannte Punktmutation gelöscht oder ausgetauscht. In manchen Fällen wird sogar eine Base hinzugefügt. Eine einzige solche Basen-Veränderung kann jedoch schon bewirken, dass der Bauplan für ein Protein fehlerhaft wird. Dies hat zur Folge, dass das Protein entweder gar nicht mehr oder in defekter Form produziert wird und das kann weitreichende Auswirkungen im Körper haben.
Ein Beispiel dafür ist die Muskeldystrophie des Typs Duchenne, an der etwa eine von 3.500 Personen leidet. Bei den Erkrankten ist der Abschnitt auf der DNA, der für das Muskelstrukturprotein Dystrophin codiert, von einer Basenmutation betroffen. Diese Mutation sorgt für eine Verschiebung des Leserasters, so dass der Bauplan für das Dystrophin-Protein nicht richtig abgelesen wird, was schlussendlich zum kompletten Verlust des Muskelproteins führt. Die Skelett- und Herzmuskulatur der Betroffenen bildet sich dadurch schon im Kindesalter stark zurück, was die Lebenserwartung deutlich verkürzt.
Typische Infektionserreger
Es kommt zwar nicht so häufig vor, doch auch nach Infektionen mit Erregern können Krankheiten entstehen, die zu den seltenen zählen. Ein bekanntes Beispiel dafür ist die Tollwut: Bei dieser einst häufigen viralen Zoonose, wird beispielsweise nach dem Biss eines infizierten Hundes ein Virus auf den Menschen übertragen. Dieser löst eine Gehirnentzündung aus, die meist tödlich verläuft. Weil heute nach solchen Bissen prophylaktisch ein passiver Impfstoff verabreicht wird, ist die Tollwut inzwischen rar geworden.
Eine weitere seltene Erkrankung ist der Botulismus, an dem Menschen nach einer Infektion mit dem Bakterium Clostridium botulinum erkranken. Die Sporen dieses Bakteriums werden hauptsächlich über verunreinigte Lebensmittel wie verdorbene Konserven oder verunreinigtem Honig aufgenommen und können das starke Nervengift Botulinumtoxin freisetzen. Dieses hemmt im Menschen den Neurotransmitter Acetylcholin und blockiert damit die Signalübertragung zwischen Nerven und Muskeln. Ohne eine Behandlung mit Gegengift führt der Botulismus in 90 Prozent der Fälle zum Tod. Bei einer schwachen Vergiftung ist die Diagnose aufgrund der vielen scheinbar zusammenhanglosen Symptomen hingegen oft schwierig.
Umwelt als ungewöhnlicher Auslöser
Im Laufe ihres Lebens stoßen Menschen nicht nur auf eine Vielzahl von Infektionserregern, sondern auch auf natürlich vorkommende oder industrielle Substanzen, die eine seltene Erkrankung auslösen können. Dazu gehören beispielsweise faserartige Minerale, besser bekannt als Asbest. Wenn die mikroskopisch kleinen Asbestfasern eingeatmet werden, können sie Mesotheliome auslösen. Diese seltene Krebsart ist schwer behandelbar und tritt inzwischen nur noch bei etwa ein bis 30 Personen von einer Millionen auf.
Auch eine Quecksilber-Vergiftung zählt heute zu den „Orphan diseases“. Nach einer Aufnahme von Quecksilber über die Lunge oder den Magen kann dies in die Blutlaufbahn gelangen und auch die Blut-Hirn-Schranke überwinden, welche normalerweise das Gehirn vor Krankheitserregern und Toxinen aus der Blutbahn schützt. Das Quecksilber-Molekül kann Proteine zerstören, indem es Bindungen aufspaltet, die für den Aufbau und die Funktion der Proteine essentiell sind. Damit können wichtige Funktionen in der Zelle nicht mehr ausgeführt werden, wobei vor allem Nervenzellen betroffen sind.
Vom Steinmann-Syndrom bis zur Progerie
Die seltensten unter den Seltenen
Auch unter den „orphan diseases“ gibt es einige Krankheiten, die in ihrer Seltenheit hervorstechen und mit zum Teil höchst außergewöhnlichen Symptomen erstaunen. Manche dieser Krankheiten betreffen nur einige hundert Menschen weltweit und bei anderen gibt es sogar nur eine Handvoll Betroffene.
Fibrodysplasia ossificans progressiva
Diese seltene Krankheit geht mit einer fortschreitenden Verknöcherung von Strukturen des Binde- und Muskelgewebes einher – Betroffenen wächst sozusagen ein zweites Skelett über das Erste. Schon die kleinsten Gewebeverletzungen führen sofort zu einer Verschlechterung des Gesundheitszustands, da sich anstatt von neuen Gewebezellen neues Knochengewebe bildet. Bisher sind weltweit etwa 800 akute Fälle bekannt.
Eine einzelne Mutation im Gen eines bestimmten Rezeptors ist verantwortlich für das gesteigerte Knochenwachstum: Durch die Mutation kommt es zu einer Überaktivierung dieser Andockstelle, die sich auf vielen Körperzellen befindet und das Wachstum sowie die Entwicklung des Skelett koordiniert.
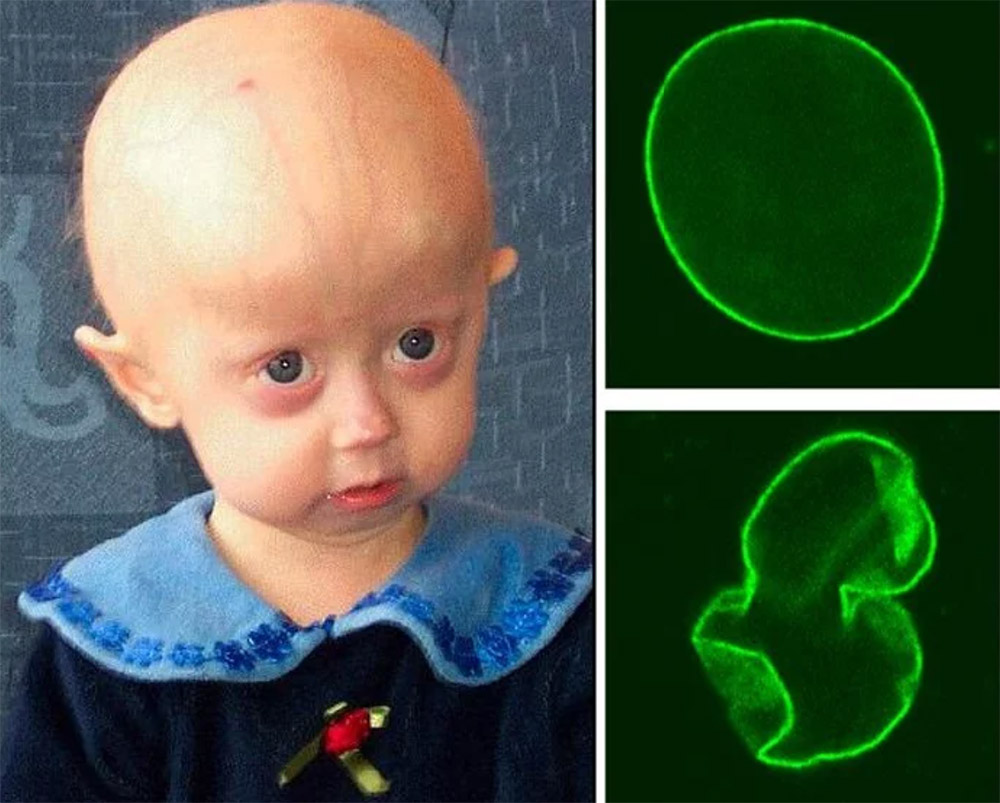
Altern wie im Zeitraffer: Das ist nicht nur eine dystopische Vorstellung, sondern für etwa 200 Kinder auf der Welt die Realität. Sie altern etwa fünf bis zehnmal schneller als normale Menschen und sterben im Alter von circa 14 Jahren meistens an einem Herzinfarkt oder einem Schlaganfall. Eine Therapie um das beschleunigte Altern aufzuhalten gibt es bisher nicht.
Die Ursachen dieser Erkrankung sind vergleichsweise gut erforscht: Eine Genmutation führt zur Produktion von Progerin, einer defekten Variante des Strukturproteins Lamin-A, welches normalerweise die Membran des Zellkerns mitaufbaut. Die schützende Membran um die Chromosomen, die die gesamte Erbinformation tragen, ist nun geschwächt und verformt sich. Dadurch können zahlreiche Prozesse der Zellteilung, wie die DNA-Replikation, nicht mehr korrekt ablaufen.
Von dieser extrem seltenen Krebsart sind seit der Erstbeschreibung 1989 nur einige Hundert Fälle weltweit bekannt. Der Tumor bildet sich vor allem im Bauchfell, welches das Innere des Bauchraums auskleidet, breitet sich in die Lymphknoten aus und streut hauptsächlich in die Leber. Bis der Tumor entdeckt wird, ist er meist schon sehr groß gewachsen und besitzt Eigenschaften, die ihn resistent gegen eine Behandlung mit Bestrahlung und Chemotherapie machen.
Die Ursache dieser seltenen Krankheit ist ebenfalls in den Genen der Betroffenen zu finden: Zwischen zwei unterschiedlichen Chromosomen findet ein Austausch eines DNA-Abschnitts statt, der ein neues Gen entstehen lässt. Dieses, so wird vermutet, ist für die Entstehung und das unkontrollierte Wachstum des Tumors verantwortlich.
Syndromale Mikrophthalmie Typ 5
Von dieser seltenen genetisch bedingten Krankheit sind bisher weniger als 20 Fälle weltweit beschrieben: Sie ist gekennzeichnet durch verschiedene Anomalien an den Augen und starken Verzögerungen der körperlichen Entwicklung. Außerdem sind Teile des zentralen Nervensystems wie der Hippocampus oder die Hypophyse fehlgebildet.
Ribose-5-Phosphat-Isomerase-Mangel
Seltener geht es nicht: Der genetisch bedingte Mangel des wichtigen Enzyms Ribose-5-Phosphat-Isomerase gilt als die seltenste bekannte Erkrankung. Bisher gibt es nur drei beschriebene Fälle von dieser schweren Stoffwechsel-Krankheit, die sich vor allem auf die weiße Substanz des Gehirns auswirkt.
Die Ribose-5-Phosphat-Isomerase spielt eine wichtige Rolle in einem Stoffwechselweg, der Teil der Umwandlung von Glucose zum chemischen Energieträger Adenosintriphosphat (ATP) ist. Die genauen molekularen Ursachen der Pathogenität sind noch nicht geklärt, doch die Patienten leiden aufgrund des fehlenden Enzyms an einer allgemeinen Entwicklungsverzögerung und haben eingeschränkte Kontrolle über ihre Bewegungen und Motorik.
Einige seltene Krankheiten erlangen weltweite Bekanntheit
Selten… und dennoch bekannt
Auf der langen Liste der seltenen Erkrankungen finden sich auch einige bekannte Namen, die man als gar nicht so selten eingeschätzt hätte. Das kann ein enormer Vorteil für die Betroffenen sein, denn mit der Bekanntheit der Krankheiten steigt auch die Wahrscheinlichkeit von finanziellen Förderungen und Investitionen in die Forschung. Aber warum sind manche Krankheiten so bekannt?
Die Krankheit Amyotrophe Lateralsklerose (ALS) tritt zwar nur bei etwa fünf von 100.000 Personen auf, aber ist dennoch den meisten Menschen ein Begriff. An dieser Erkrankung litt der berühmte Astrophysiker Stephen Hawking, der einen bedeutenden wissenschaftlichen Beitrag zur Allgemeinen Relativitätstheorie und dem Verständnis schwarzer Löcher leistete.
Bei dieser neurodegenerativen Erkrankung verlieren die motorischen Nervenzellen fortschreitend ihre Funktion, so dass die Muskulatur nicht mehr gesteuert werden kann und als Folge davon abgebaut wird. Die Mund- und Zungenmuskulatur ist beispielsweise auch betroffen, wodurch die Patienten ab einem bestimmten Stadium nicht mehr sprechen können.
Die Bekanntheit der Nervenkrankheit gipfelte im Sommer 2014 in der „Ice Bucket Challenge“. Bei dieser Spendenkampagne sollte man sich in einem Video einen Eimer mit Eiswasser über den Kopf schütten, weitere Personen nominieren und anschließend zehn US-Dollar oder Euro an die ALS Association (ALSA) spenden. Dies entwickelte sich in den sozialen Medien zu einem echten Hype, und auch wenn irgendwann der tatsächliche Sinn der Aktion etwas in den Hintergrund geriet, gingen im Jahr 2014 allein innerhalb einer Augustwoche 41,8 Millionen Dollar bei der ALSA ein. Zum Vergleich: Im selben Zeitraum des Vorjahres erhielt die Organisation 2,1 Millionen Dollar Spendengelder.
Wer kein weltberühmter Physiker ist, kann auch anderweitig die eigene Krankheitsgeschichte erzählen, wie das Beispiel Susannah Cahalans zeigt. In ihrem Buch berichtet sie die unglaubliche Geschichte von einer Immunerkrankung, die sie selbst als „Brain on fire“ bezeichnet und die viele Ärzten als Schizophrenie abtaten.
Cahalan litt unter plötzlichen Verhaltensveränderungen, Migräne, Bewusstseinsstörungen und epileptischen Anfällen. Von Depressionen über Alkoholentzugserscheinungen bis hin zur Schizophrenen Psychose erhielt die Amerikanerin viele Fehldiagnosen, bis schließlich ein Arzt ihre extrem seltene Erkrankung erkannte: Anti-NMDA-Rezeptor-Enzephalitis. Bei dieser seltenen Autoimmunerkrankung bildet der Körper Antikörper gegen den NMDA-Rezeptor, der eine wichtige Rolle bei der Signalübertragung im Gehirn spielt. Der Körper greift dadurch das eigene Gehirn an.
Mehr Aufmerksamkeit schaffen
Cahalan ist erst die 217. Patientin, bei der Ärzte diese Krankheit nach der Erstbeschreibung im Jahr 2007 diagnostizierten. Spätestens nachdem ihre Krankheitsgeschichte mit „Brain of fire“ im Jahr 2016 verfilmt wurden, erlangte ihre Geschichte und damit auch ihre Krankheit internationale Bekanntheit. Mit ihrer Erzählung will Cahalan ihre seltene Krankheit mehr in den Fokus rücken: „Wegen des Artikels, den ich geschrieben, konnte ich die Nachricht verbreiten. Menschen wurden diagnostiziert, aufgrund dessen, was ich geschrieben habe“, sagte Cahalan gegenüber der britischen Zeitung „The Guardian“.
Auch die Ärztin Lisa Sanders nutzt soziale Netzwerke und Massenmedien, um das Bewusstsein für seltene Erkrankungen zu erhöhen. „Es ist wichtig, jeder Person, die eine Diagnose bekommen kann, auch eine zu ermöglichen“, sagt Sanders. Die Professorin an der Yale University schreibt seit 2002 für die New York Times die Kolumne „Diagnosis“, in der sie rätselhafte Krankheitsbilder ihrer Patienten vorstellt. Ihre große Reichweite will sie im Sinne des „Crowdsourcing“ nutzen, um mit dem „Wissen der Masse“ die seltenen Symptome diagnostizieren zu können. Gleichzeitig soll die Kolumne anderen Betroffenen vermitteln, dass sie mit ihrer Erkrankung nicht alleine sind.
Wie können seltene Erkrankungen behandelt werden?
Gentherapien für Orphan Diseases
Das schnell wachsende Feld der Gentherapien bietet besonders bei den seltenen Erkrankungen neue Chancen. Denn ein Großteil der „Orphan diseases“ sind auf ein einzelnes Gen zurückzuführen und Gentherapeutika können diese Defekte im Genom der Betroffenen reparieren. Typischerweise wird dafür Genmaterial mit der korrigierten DNA-Abfolge durch eine virale „Genfähre“ in das Erbgut des Betroffenen eingeschleust.
Viren gelten als gut geeignete Gen-Taxis, denn sie sind von Natur aus darauf ausgerichtet, ihr Erbgut in Zellen einzuschleusen. Dafür docken sie an die Oberfläche von menschlichen Zellen an und injizieren ihr Erbgut. Daraufhin fangen die Zellen an, die in der Erbsubstanz codierten Proteine zu produzieren, was sich wiederum die Entwickler von Gentherapien zu Nutze machen: Sie entfernen alle für den Menschen schädlichen Gene des Virus und ersetzen sie durch den „gesunden“ DNA-Abschnitt, der den Patienten sonst fehlt.
EU schafft Anreize für Pharmaunternehmen
Um die Entwicklung von Therapien gegen seltene Erkrankungen voranzutreiben, verabschiedete die Europäische Union im Jahr 2000 die Orphan Drug-Verordnung. Damit sollten wirtschaftliche Anreize für das Design solcher Medikamente geschaffen werden.
Beispielsweise sind Pharmaunternehmen für zehn Jahre vor sogenannten Nachahmer-Präparaten geschützt. Das bedeutet, dass Medikamente nicht für die gleiche Krankheit zugelassen werden können, wenn sie nicht signifikant besser wirken und weiteren medizinischen Fortschritt bringen. Somit können sich Unternehmen ein Alleinstellungs-Merkmal verschaffen. Ein weiterer wirtschaftlicher Anreiz durch diese Verordnung wird durch reduzierte Gebühren für die Zulassung und die wissenschaftliche Beratung durch die Zulassungsbehörde EMA geschaffen.
Die Beantragung des Orphan-Drug-Status bringt für Pharmaunternehmen also einige Vorteile. Dies führte seit 2000 zu einem deutlichen Anstieg der Zulassung für Medikamente gegen seltene Erkrankungen: Während die Forschung vor dem Jahr 2000 etwa ein Medikament pro Jahr herausbrauchte, liegt die Zahl der pro Jahr zugelassenen Medikamente mittlerweile im zweistelligen Bereich.
Die Zwei Millionen Dollar-Frage
Aktuell sind in der EU für 143 seltene Erkrankungen Medikamente zugelassen, darunter beispielsweise den schwere Immundefekt ADA-SCID, die Stoffwechselerkrankung Alpha-Mannosidase oder die Muskelkrankheit Duchenne Muskeldystrophie.
Am 18. Mai 2020 erhielt die Gentherapie Zolgensma gegen Spinale Muskelatrophie von der Europäischen Arzneimittelagentur (EMA) eine Zulassung. Mit einer einzigen Infusion kann damit die lebendsbedrohliche Krankheit, von der in Deutschland jährlich etwa 80 Kinder betroffen sind, geheilt werden. Allerdings geraten Eltern und Krankenkassen unter Druck: Mit 1,945 Millionen Euro ist die Gentherapie Zolgensma das bislang teuerste Medikament der Welt.
Wie kommen solche horrenden Preise zu Stande? Nach Angaben der Pharmaindustrie belaufen sich die Kosten für die Entwicklung eines solchen Medikaments auf etwa zwei Milliarden US-Dollar. Das liegt unter anderem daran, dass 90 Prozent aller Versuche, ein neues Medikament zu entwickeln, fehlschlagen. Es müssen also auch die Kosten für die zahlreichen misslungenen Medikamente abgedeckt werden.
Außerdem gibt es in der Regel wenig Kunden: Damit sich die Entwicklung einer Gentherapie für die Hersteller lohnt, muss das Medikament bei nur einigen hundert potenziellen Abnehmern etwa eine Million US-Dollar kosten. Und die Pharmaunternehmen, wissend um die Verzweiflung der Eltern eines Kindes mit Gendefekt, verlangen auch solche Preise.
Wie stemmen das die Krankenkassen?
In Deutschland werden die Kosten der Gentherapien bei einem nachgewiesenen medizinischen Nutzen in den meisten Fällen von den Krankenkassen übernommen, so dass das finanzielle Risiko bei den Kassen liegt. Vor allem unter dem Gesichtspunkt der steigenden Anzahl von Zulassungen der teuren Gentherapien verlangen deshalb nun viele Krankenkassen nach einem neuen Erstattungsmodell.
Da die Behandlung mit Gentherapien meist einmalig erfolgt, müssen die Krankenkassen den hohen Preis der Medikamente meist auch auf einen Schlag bezahlen. Eine gestaffelte Bezahlung des Preises über Jahre könnte hier eine Lösung sein. Eine weiteres alternatives Erstattungskonzept sieht erfolgsabhängige Modelle vor, auch genannt „Pay-for-performance“. Da oft Langzeitstudien fehlen und nicht genau klar ist, ob die Therapie ein Leben lang wirkt, sollen bis zu einem bestimmten Zeitpunkt Erfolge bei der Behandlung nachgewiesen werden. Wirkt die Therapie nicht so wie gedacht, verpflichtet sich das jeweilige Pharmaunternehmen zu Rückzahlungen.
Ob sich die Modelle durchsetzen, bleibt abzusehen. Doch um die Zukunft der Heilung von seltenen Erkrankungen mit Gentherapeutika nicht zu gefährden, müssen Hersteller und Krankenkassen eine Lösung finden.