Prionkrankheiten wie die Creutzfeldt-Jakob-Krankheit treten beim Menschen meist spontan. Typisch dafür sind Ablagerungen einer falsch gefalteten Form (PrPSc) des zellulären Prionproteins (PrPC). Was die fehlerhafte Eiweißfaltung auslösen kann, war bisher weitgehend unbekannt. Jetzt aber haben Wissenschaftler gezeigt, dass eine Oxidation im Inneren von PrPC den Startschuss für die fatale Umstrukturierung geben kann.
Da auch andere neurodegenerative Erkrankungen auf der Fehlfaltung von Proteinen beruhen, könnten diese Ergebnisse helfen, auch die Entstehungsmechanismen beispielsweise von Alzheimer und Parkinson besser zu verstehen, so die Forscher des Max-Planck-Instituts für Biochemie und der Universität München (LMU) in der Early Edition der „Proceedings of the National Academy of Sciences“ (PNAS).
Ziehharmonika-ähnlich gefaltete Proteinbänder
PrPC ist im Zentralnervensystem weit verbreitet. Als Strukturelemente enthält es vor allem spiralförmige alpha-Helices. Die fehlerhafte Faltung dieser alpha-Helices in ziehharmonika-ähnlich gefaltete Proteinbänder – so genannte beta-Faltblattstrukturen – verändert die Eigenschaften des Proteins: Es wird schlechter wasserlöslich und neigt dazu, mit weiteren Proteinmolekülen zu aggregieren. Es kommt zur Bildung unlöslicher Protein-Klumpen, die sich in den Zellen ablagern und diese schädigen – ein Charakteristikum der Prionkrankheiten.
„Ist erst einmal fehlgefaltetes und aggregiertes Prionprotein im Gewebe vorhanden, wird eine Kettenreaktion ausgelöst, in der sich die Umfaltung der Prionproteine fortpflanzt, wie bei Dominosteinen, die sich gegenseitig umstoßen, sobald der erste Stein gekippt ist“, erklärt Professor Armin Giese vom Zentrum für Neuropathologie und Prionforschung der LMU.
Prionkrankheiten können auf verschiedenen Wegen ausgelöst werden: Es gibt familiäre Formen, die auf Genmutationen beruhen, oder man kann sich mit infiziertem Material anstecken. Die meisten Fälle aber entstehen beim Menschen vermutlich durch die spontane Umstruktierung der alpha-Helices in beta-Faltblätter. Was diese spontane Umwandlung auslöst, war bisher unbekannt.
Neuer Erklärungsansatz gefunden
Nun haben die Münchener Wissenschaftler einen Erklärungsansatz gefunden: „Zwar werden auch andere Mechanismen für die spontane Umwandlung in die bösartige Variante diskutiert, unserer Ansicht nach spielt aber die Oxidation der Aminosäure Methionin im Inneren des Prionproteins eine entscheidende Rolle“, berichtet Dr. Nediljko Budisa vom Max-Planck-Institut für Biochemie.
Methionine kommen in PrPC an mehreren Stellen vor: Einige sitzen auf der Außenseite des Moleküls, zwei dagegen befinden sich gut abgeschirmt im Inneren der Molekülstruktur. Methionin wird durch reaktive Sauerstoffradikale leicht oxidiert, normalerweise erreicht der reaktive Sauerstoff aber nur die gut zugänglichen Methionine auf der Moleküloberfläche. Dort sorgt das Enzym Methionin-Sulfoxidreduktase dafür, dass oxidierte Methionine auch wieder reduziert werden.
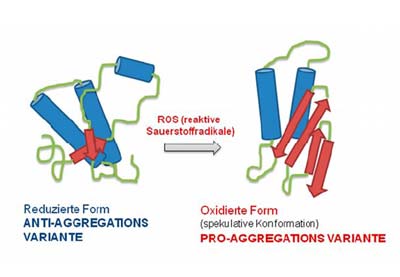
Gefährlich wird es, wenn dieses Entgiftungsenzym nicht ausreichend funktioniert – „z.B. im Alter arbeiten Enzyme manchmal nicht mehr so gut“, erklärt Budisa – oder die Zelle in oxidativen Stress gerät: Stehen sehr viele Sauerstoffradikale zur Verfügung, werden auch die Methionine im Molekülinneren oxidiert – ein irreversibler Prozess, der Folgen hat: Während das recht hydrophobe Methionin normalerweise Helixstrukturen effektiv stabilisiert, unterstützt seine oxidierte Form die Umfaltung in Faltblattstrukturen. „Durch die Oxidation wird das Prionprotein auseinandergedrückt und platzt regelrecht, das ist für die Faltung natürlich verheerend“, sagt Budisa.
Wissenschaftler packen Tricks aus
Eine künstliche Oxidation aller Methionine unter kontrollierten Bedingungen in der Zelle scheitert experimentell an der effizienten Arbeit der Methionin-Sulfoxidreduktase, deshalb war der endgültige Beweis, dass das Aggregieren der Prione tatsächlich mit der Methioninoxidation zusammenhängt, schwierig zu führen. Die Wissenschaftler lösten dieses Problem mit einem eleganten Trick: Sie erzeugten maßgeschneiderte Prionproteine, bei denen Methionin durch seine Analoge Norleucin beziehungsweise Methoxinin ersetzt ist. Diese Analoge sind chemisch stabil, werden nicht weiter oxidiert und simulieren in ihren chemischen Eigenschaften Methionin (Norleucin) beziehungsweise dessen oxidierte Form (Methoxinin).
„Dadurch werden künstliche Prionproteine geschaffen, die wie Yin und Yang zwei extreme Zustände widerspiegeln“, erklärt Budisa: Ein Prion, in dem nur nicht oxidierte Methionine vorhanden sind, und eines, in dem alle Methionine oxidiert wurden. Dabei zeigte sich: Mit Norleucin resultiert ein Prionprotein, das reich an Helixstrukturen ist – wie das natürliche Prionprotein – und dessen Aggregationsneigung gering ist. Mit Methoxinin dagegen resultierten Prionproteine, die reich an Faltblättern sind und stark aggregieren. Im Gegensatz zum Ursprungs-Prionprotein sind beide künstliche Varianten unempfindlich gegenüber weiterer Oxidation durch freie Sauerstoffradikale – ihre unterschiedliche Struktur verdanken sie demnach den verschiedenen Methionin-Substituten.
Wichtige Ergebnisse auch für Alzheimer und Parkinson
Die Ergebnisse zeigen nach Angaben der Forscher klar einen Zusammenhang zwischen oxidativem Stress in der Zelle und der Fehlfaltung von Proteinen. Dies ist nicht nur für die Prionforschung und die damit zusammenhängenden Erkrankungen hoch interessant, sondern auch für andere neurodegenerative Erkrankungen die mit Fehlfaltungen von Proteinen zu tun haben, wie Alzheimer und Parkinson.
Vor dem Hintergrund einer alternden Gesellschaft wird die Zahl der Patienten mit neurodegenerativen Erkrankungen, die sich meist erst im höheren Alter manifestieren, noch zunehmen. Die Forschung auf diesem Gebiet kann einen Beitrag leisten, diese altersbedingten Erkrankungen besser zu verstehen und möglicherweise neue Therapiestrategien zu entwickeln.
(idw – Max-Planck-Institut für Biochemie, 22.04.2009 – DLO)