Das Gen, das für die Entstehung einer seltenen erblichen Variante der Parkinson-Krankheit verantwortlich ist, hat jetzt ein internationales Wissenschaftler-Team in einer Studie identifiziert. Das PARK9 oder Kufor-Rakeb Syndrom (KRS), kann bereits in der Jugend auftreten. Betroffene zeigen neben typischen Zeichen einer Parkinson-Erkrankung eine ausgeprägte Neurodegeneration einschließlich Demenz.
{1l}
Die Forscher um Professor Christian Kubisch von der Universität zu Köln untersuchten in Zusammenarbeit mit Arbeitsgruppen aus Marburg, Bonn, England, Chile und Jordanien zwei Familien, in denen das Kufor-Rakeb Syndrom bei mehreren Mitgliedern aufgetreten ist. Dabei entdeckten sie das Gen ATP13A2. Beide Kopien dieses Gens sind bei erkrankten Familienmitgliedern von Mutationen betroffen. Jede dieser Genveränderungen führt zu einem Verlust der Funktion des Erbgutbausteins, so die Forscher in der Oktober-Ausgabe des renommierten Wissenschaftsjournals "Nature Genetics".
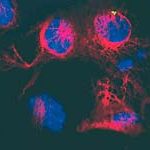
Das Gen ATP13A2 kodiert für ein Protein, das in der Membran von so genannten Lysosomen vorkommt. Das sind kleine Zellorganellen, die Proteine aufnehmen und abbauen. Das ATP13A2 Protein findet sich überall im Körper. Am stärksten wird es jedoch im Gehirn gebildet, vor allem in der Substantia nigra, einer Region von der bekannt ist, dass sie eine zentrale Rolle bei der Parkinson-Krankheit spielt.
Da bei KRS-Patienten beide Kopien des Gens verändert sind, kann kein funktionales Protein gebildet werden. Nach ersten Erkenntnissen der Wissenschaftler wird das mutierte Protein nicht in die Membran der Lysosomen eingebaut, sondern in einem weiteren Zellorganell, dem endoplasmatischen Reticulum, als fehlerhaft erkannt und nachfolgend von der Zelle abgebaut. Dies könnte zu einem verringerten Abbau anderer Proteine führen, was wiederum eine toxische Anhäufung zur Folge hätte.
Neue Studien geplant
Aber auch eine Funktionseinschränkung der Lysosomen selbst könnte durch die Anhäufung von abzubauenden Proteinen auftreten. "Solche Störungen in Abbausystemen der Zelle sind eine bekannte Ursache von neurodegenerativen Erkrankungen", erläutert Kubisch. "Mit ATP13A2 haben wir nun eine neue Klasse von Proteinen entdeckt, die eine Rolle bei der Entstehung von neurodegenerativen Erkrankungen und insbesondere der Parkinson-Erkrankung spielen könnten."
Interessanterweise konnten die Wissenschaftler zeigen, dass dieses Gen auch im Gehirn von Patienten mit typischem Morbus Parkinson differenziell reguliert ist, sodass erste Hinweise darauf existieren, dass ATP13A2 nicht nur beim seltenen Kufor-Rakeb-Syndrom sondern eventuell auch bei den häufigen neurodegenerativen Erkrankungen beteiligt ist. Diese Fragestellung und die Aufklärung der eigentlichen Funktion des ATP13A2 Proteins stehen im Zentrum der geplanten nachfolgenden Studien der Wissenschaftler.
(Nationales Genomforschungsnetz NGFN, 22.09.2006 – DLO)